Overview
The pathology processing pipeline in HoneyBee handles Whole Slide Images (WSIs), which are high-resolution scans of tissue samples. These images present unique computational challenges due to their extreme size (often several gigabytes), multi-resolution pyramid structure, and vendor-specific file formats. HoneyBee uses a four-class modular design — Slide → PatchExtractor → Patches → PathologyProcessor — so each stage can be used independently or composed into end-to-end pipelines.
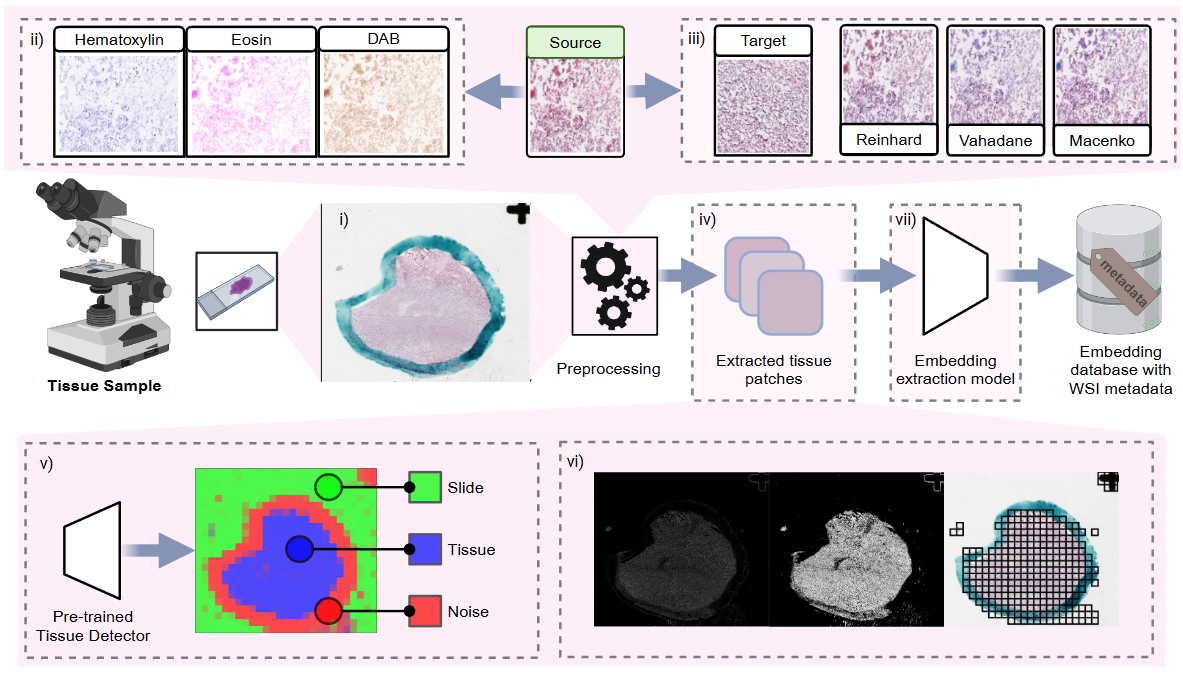
Key Features
- Support for multiple WSI formats (Aperio SVS, Philips TIFF, and more)
- Auto-detecting backend: CuCIM (GPU-accelerated) with OpenSlide fallback
- Deep-learning and classical tissue detection (Otsu, HSV, gradient)
- Stain normalization (Reinhard, Macenko, Vahadane) and H&E stain separation
- Grid-based patch extraction with tissue filtering and quality scoring
- 8 foundation model presets (UNI, UNI2, Virchow2, H-optimus, GigaPath, Phikon-v2, MedSigLIP, REMEDIS)
- Slide-level aggregation (mean, max, median, std, concat)
- Built-in visualizations: tissue masks, patch galleries, quality distributions, UMAP feature maps
Quick Start
Install HoneyBee with pathology dependencies and download a sample slide from HuggingFace:
import torch
from huggingface_hub import hf_hub_download
from honeybee.loaders.Slide.slide import Slide
from honeybee.processors import PathologyProcessor
from honeybee.processors.wsi import PatchExtractor
# Download a sample WSI from HuggingFace
SLIDE_PATH = hf_hub_download(
repo_id="Lab-Rasool/honeybee-samples",
filename="sample.svs",
repo_type="dataset",
)
DEVICE = "cuda" if torch.cuda.is_available() else "cpu"Slide Loading
The Slide class auto-detects the best available backend
(CuCIM for GPU acceleration, OpenSlide as fallback) and provides a
unified API for reading WSI files. Use slide.info to
inspect metadata and slide.dimensions for the full-resolution size.
slide = Slide(SLIDE_PATH)
# Slide metadata (backend, dimensions, level count, magnification, etc.)
print(slide.info)
# Full-resolution dimensions (width, height)
print(slide.dimensions) # e.g. (27965, 25146){
"path": "sample.svs",
"backend": "cucim",
"dimensions": [27965, 25146],
"level_count": 3,
"level_dimensions": [[27965, 25146], [6991, 6286], [1747, 1571]],
"level_downsamples": [1.0, 4.0, 16.0],
"magnification": null,
"mpp": 1.0,
"vendor": null
}Thumbnails and Region Reading
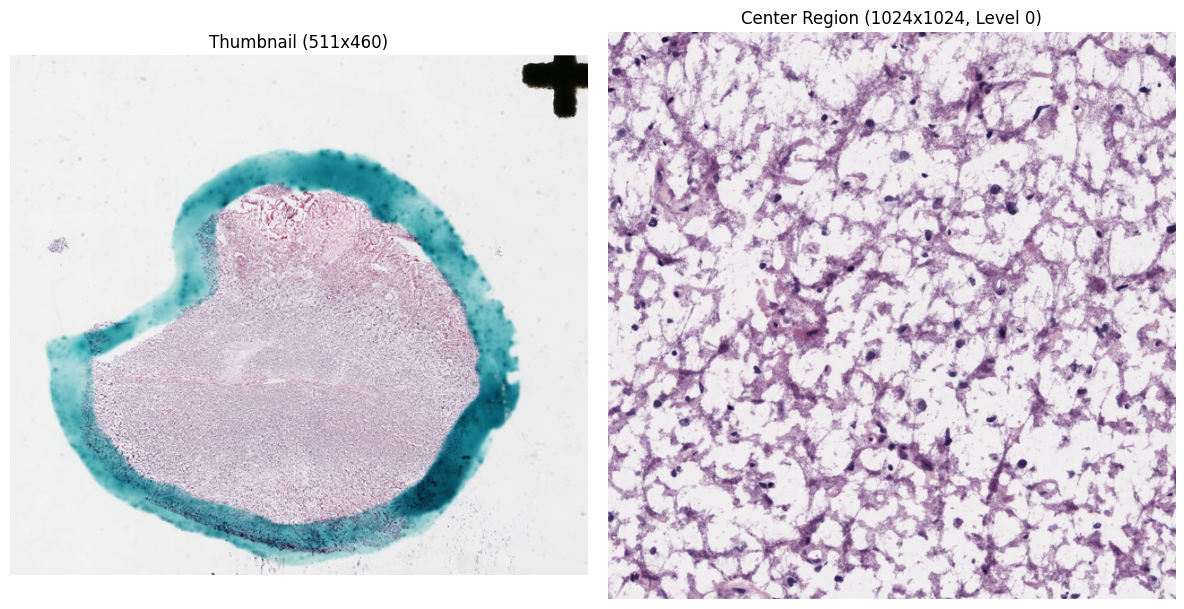
get_thumbnail() returns a downsampled overview of the entire slide.
read_region() reads pixels at a specific level-0 location and size,
returning an RGB NumPy array.
# Downsampled overview
thumbnail = slide.get_thumbnail(size=(512, 512))
# Read a 1024x1024 region from the center of the slide
cx, cy = slide.dimensions[0] // 2, slide.dimensions[1] // 2
region = slide.read_region(
location=(cx - 512, cy - 512),
size=(1024, 1024),
level=0,
)
Tissue Detection
HoneyBee provides both deep-learning and classical approaches for
tissue detection. Results are stored on the slide object as
slide.tissue_mask and slide.prediction_map.
Deep Learning Detection
Uses a pretrained DenseNet121 model for fine-grained tissue segmentation.
The patch_size parameter controls tile resolution —
smaller patches yield finer masks at the cost of more inference passes.
slide.detect_tissue(
method="dl",
device=DEVICE,
patch_size=64,
thumbnail_size=(4096, 4096),
)
print(f"Tissue mask: {slide.tissue_mask.shape}")
print(f"Tissue ratio: {slide.tissue_mask.mean():.2%}")
print(f"Prediction map: {slide.prediction_map.shape}")
# Visualize the detection result
slide.plot_tissue_detection()Tissue mask: (3683, 4095), ratio: 29.48%
Prediction map: (24, 27, 3)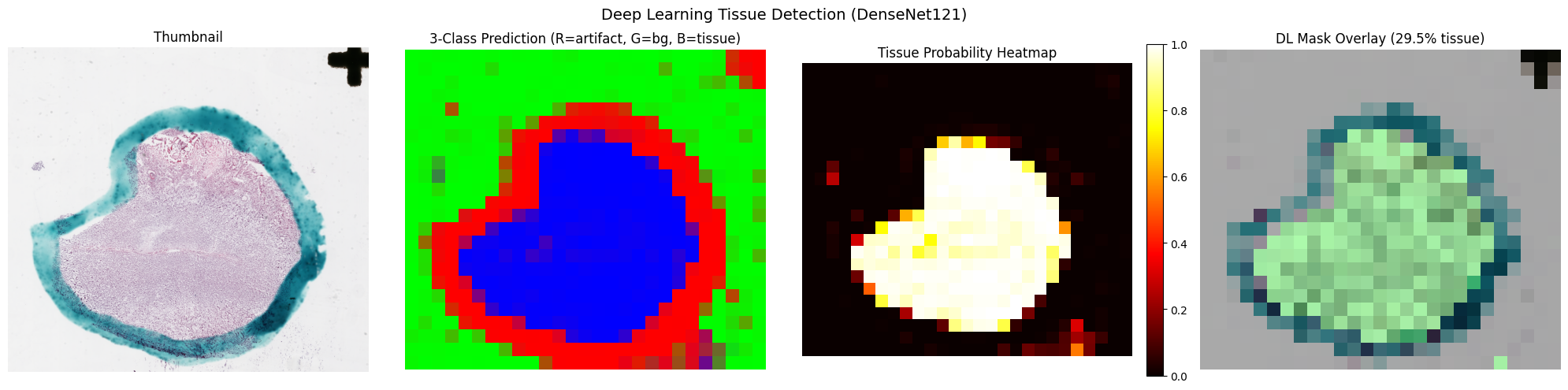
Classical Methods
Three classical approaches are available: Otsu thresholding
("otsu"), HSV color filtering ("hsv"),
and their combination ("otsu_hsv").
# Otsu thresholding
slide.detect_tissue(method="otsu")
# HSV color filtering
slide.detect_tissue(method="hsv")
# Combined Otsu + HSV
slide.detect_tissue(method="otsu_hsv")Method Comparison
Compare detection methods side-by-side to choose the best fit for your data:
slide.compare_tissue_methods(["dl", "otsu", "hsv", "otsu_hsv"])
Patch Extraction
PatchExtractor performs grid-based extraction using the slide's
tissue mask to filter out background tiles. Configure patch size, stride,
and minimum tissue ratio to control density.
Grid Preview
Visualize the extraction grid over the tissue mask before committing to pixel reads:
extractor = PatchExtractor(
patch_size=256,
stride=256,
min_tissue_ratio=0.5,
)
# Preview the grid overlay on the tissue mask
extractor.plot_grid_preview(slide)
Extract and Inspect
extract() returns a Patches container holding
image arrays and coordinates. Use built-in visualizations to inspect results.
patches = extractor.extract(slide)
print(f"Extracted {len(patches)} patches")
print(f"Images shape: {patches.images.shape}")
print(f"Coordinates shape: {patches.coordinates.shape}")
# Gallery of extracted patches
patches.plot_gallery(cols=8, max_patches=64)
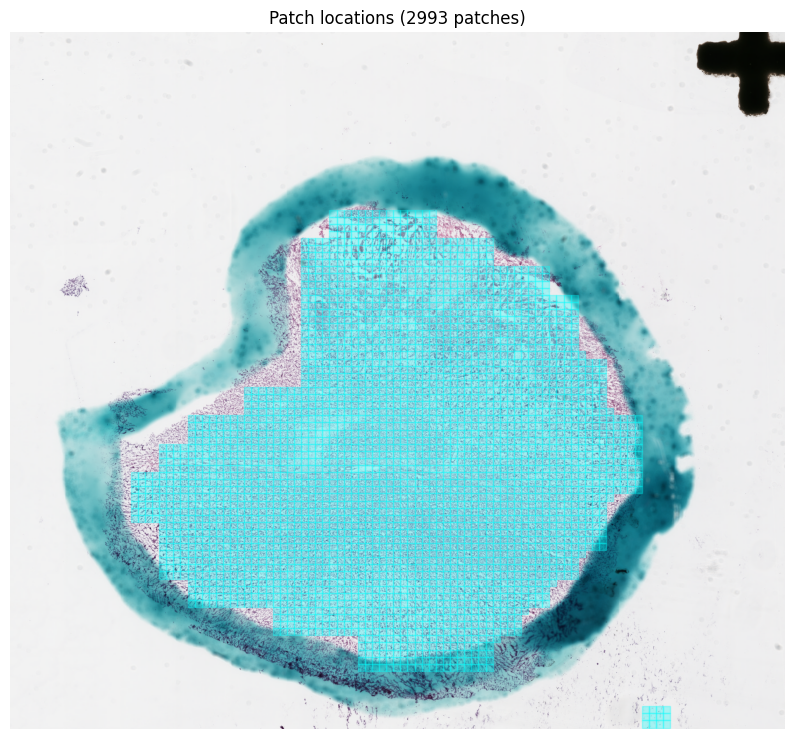
# Patch locations overlaid on the slide thumbnail
patches.plot_on_slide(slide)Extracted 2993 patches
Images shape: (2993, 256, 256, 3)
Coordinates shape: (2993, 4)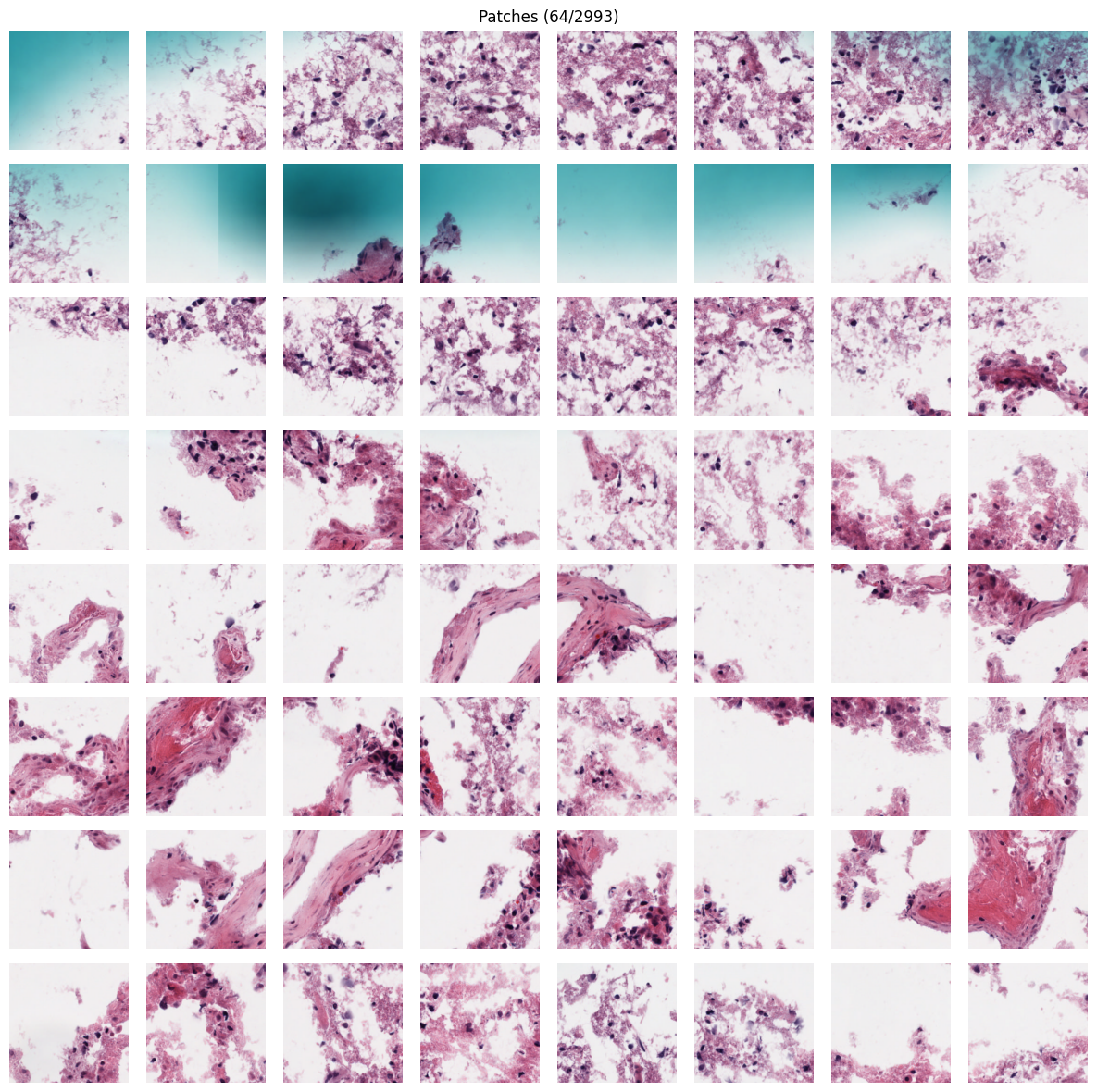
Quality Filtering
Quality scores combine tissue ratio, color variance, and edge content.
Use plot_quality_distribution() to choose a threshold, then
filter() to discard low-quality patches. The Patches
container is immutable — filtering returns a new instance.
# Visualize quality score distribution with a threshold line
patches.plot_quality_distribution(threshold=0.7)
# Filter patches (returns a new Patches instance)
good_patches = patches.filter(min_quality=0.7)
print(f"Filtered: {len(patches)} -> {len(good_patches)} patches")Filtered: 2993 -> 249 patches (min_quality=0.7)Multi-Resolution Extraction
For large slides, run tissue detection at low magnification to build a
coarse spatial grid, then pass it to a high-resolution extractor via the
tissue_coordinates parameter. This avoids re-running
detection at full resolution.
# Low-res tissue grid: small patches at coarse magnification
lowres_extractor = PatchExtractor(
patch_size=16, stride=16, magnification=5.0, min_tissue_ratio=0.3
)
tissue_grid = lowres_extractor.get_coordinates(slide)
print(f"Low-res tissue grid: {len(tissue_grid)} tiles")
# High-res extraction using the tissue grid as a spatial filter
hires_extractor = PatchExtractor(
patch_size=256, stride=256, magnification=20.0, min_tissue_ratio=0.5
)
hires_patches = hires_extractor.extract(slide, tissue_coordinates=tissue_grid)
print(f"High-res patches: {len(hires_patches)}")Low-res tissue grid: 809280 tiles (16px @ ~5x)
High-res patches: 3188 (256px @ ~20x)
Tissue filter: tissue_coordinates
Tissue coordinates used: 809280
Stain Normalization
Stain normalization reduces color variability across slides from different
scanners and labs. Three methods are available: Reinhard, Macenko, and Vahadane.
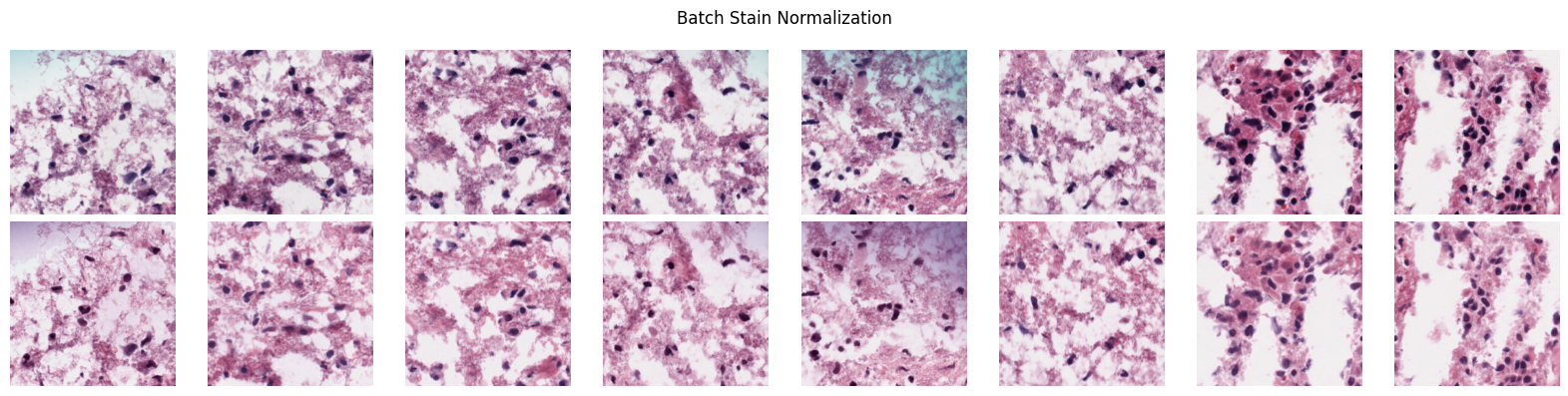
All stain operations on Patches are immutable and return new instances.
# Compare all normalization methods side-by-side on a single patch
good_patches.plot_normalization_comparison()
# Apply Macenko normalization (returns a new Patches instance)
normalized = good_patches.normalize(method="macenko")
# Before/after visualization
good_patches.plot_normalization_before_after(normalized)Normalized 249 patches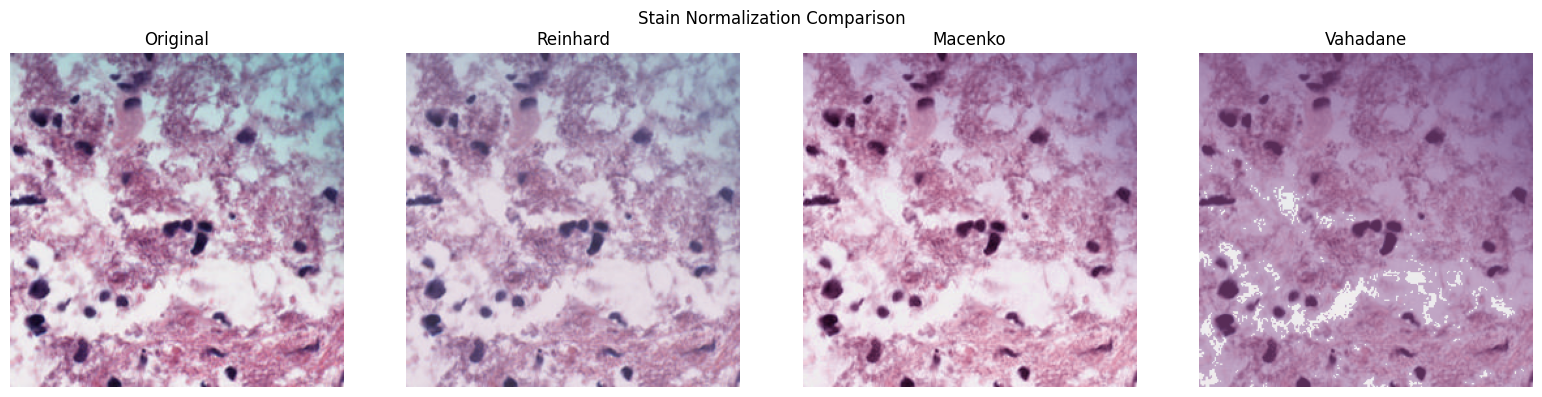
H&E Stain Separation
Deconvolve patches into hematoxylin, eosin, and background channels using color deconvolution:
patches.plot_stain_separation()
Embedding Generation
PathologyProcessor wraps the model registry to generate
patch-level embeddings from any supported foundation model. Pass a
Patches object directly to generate_embeddings().
Available Models
HoneyBee ships with 8 preset foundation models. Use list_models()
to see all available presets, or pass any HuggingFace / timm model ID with an
explicit provider.
| Alias | Embedding Dim | Provider | Description |
|---|---|---|---|
uni | 1024 | timm | UNI ViT-L/16 pathology foundation model (MahmoodLab) |
uni2 | 1536 | timm | UNI2-h ViT-H/14 pathology foundation model (MahmoodLab) |
virchow2 | 2560 | timm | Virchow2 ViT-H/14 pathology model (Paige AI) - cls+mean pooling |
h-optimus | 1536 | timm | H-optimus-0 pathology foundation model (Bioptimus) |
gigapath | 1536 | timm | Prov-GigaPath DINOv2-based pathology model |
phikon-v2 | 1024 | huggingface | Phikon-v2 pathology foundation model (Owkin) |
medsiglip | 1152 | huggingface | MedSigLIP medical image-text model (Google) - 448x448 |
remedis | 2048 | onnx | REMEDIS CXR model (Google) - requires ONNX model_path |
from honeybee.models.registry import list_models
# List all registered model presets
for m in list_models():
print(f" {m['alias']:>12s} dim={m['embedding_dim']:>4d} provider={m['provider']}") gigapath dim=1536 provider=timm
h-optimus dim=1536 provider=timm
medsiglip dim=1152 provider=huggingface
phikon-v2 dim=1024 provider=huggingface
remedis dim=2048 provider=onnx
uni dim=1024 provider=timm
uni2 dim=1536 provider=timm
virchow2 dim=2560 provider=timmGenerate Embeddings
Initialize PathologyProcessor with a model alias, then call
generate_embeddings() with a Patches object:
processor = PathologyProcessor(model="uni2")
# Inspect model configuration
info = processor.get_model_info()
print(f"Model: {info['alias']}, dim: {info['embedding_dim']}")
# Generate patch-level embeddings
embeddings = processor.generate_embeddings(
patches,
batch_size=32,
progress=True,
)
print(f"Embeddings shape: {embeddings.shape}") # (num_patches, embedding_dim)Model: uni2, dim: 1536
Embeddings shape: (2993, 1536)Slide-Level Aggregation
Aggregate patch-level embeddings into a single slide-level representation using one of five methods:
# Available methods: mean, max, median, std, concat
for method in ["mean", "max", "median", "std", "concat"]:
agg = processor.aggregate_embeddings(embeddings, method=method)
print(f" {method:>8s}: shape={agg.shape}") mean: shape=(1536,)
max: shape=(1536,)
median: shape=(1536,)
std: shape=(1536,)
concat: shape=(3072,)UMAP Feature Maps
Project high-dimensional embeddings to 3D with UMAP, map each dimension to an RGB channel, and overlay on the slide thumbnail. Similar tissue regions receive similar colors.
processor.plot_feature_map(patches, embeddings, slide)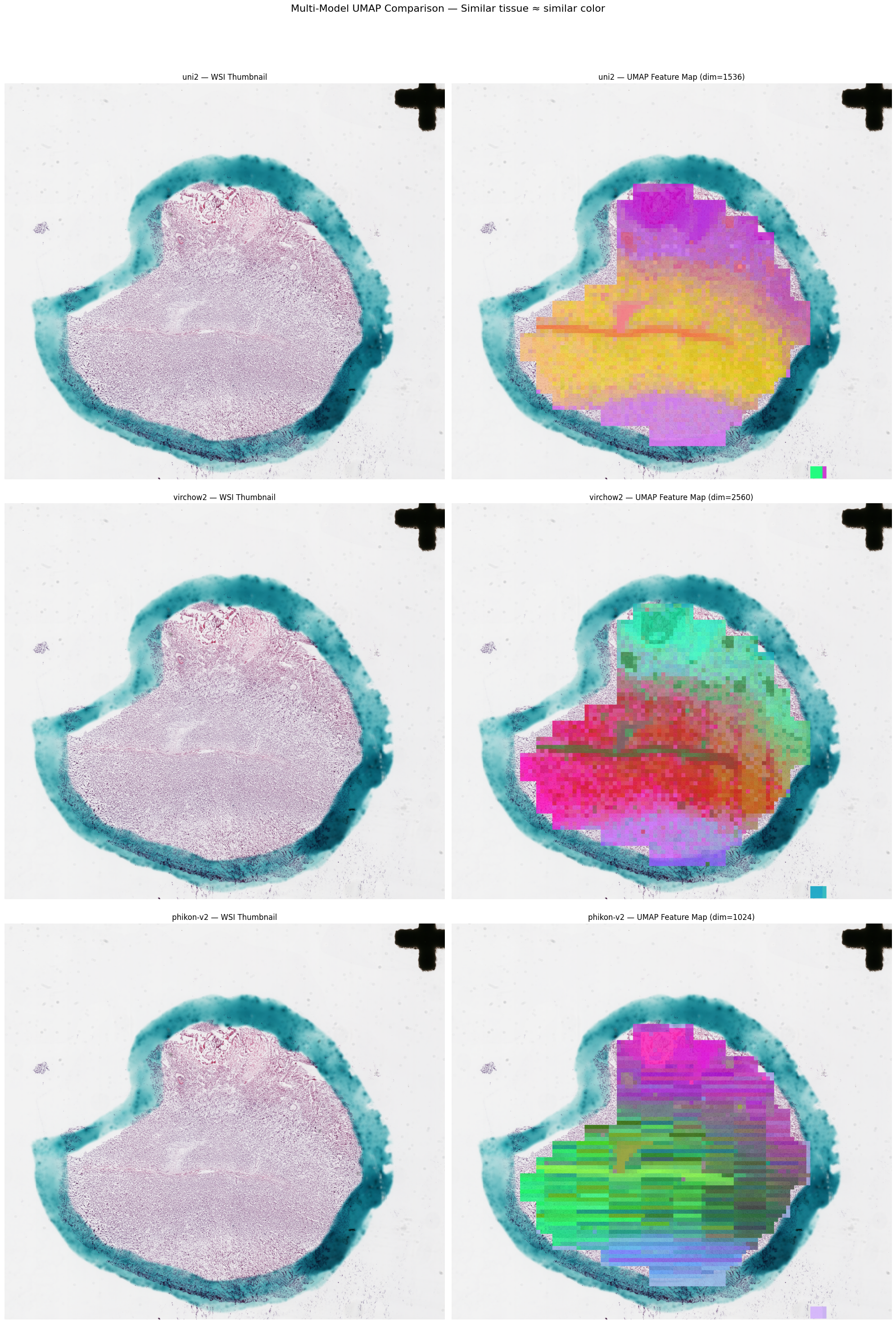
Complete Pipeline Example
Full end-to-end workflow from WSI loading to slide-level embeddings:
import torch
from huggingface_hub import hf_hub_download
from honeybee.loaders.Slide.slide import Slide
from honeybee.processors import PathologyProcessor
from honeybee.processors.wsi import PatchExtractor
# 1. Load slide
slide_path = hf_hub_download(
repo_id="Lab-Rasool/honeybee-samples",
filename="sample.svs",
repo_type="dataset",
)
slide = Slide(slide_path)
# 2. Detect tissue
device = "cuda" if torch.cuda.is_available() else "cpu"
slide.detect_tissue(method="dl", device=device, patch_size=64)
# 3. Extract patches
extractor = PatchExtractor(patch_size=256, stride=256, min_tissue_ratio=0.5)
patches = extractor.extract(slide)
# 4. Quality filtering
good_patches = patches.filter(min_quality=0.7)
# 5. Stain normalization
normalized = good_patches.normalize(method="macenko")
# 6. Generate embeddings
processor = PathologyProcessor(model="uni2")
embeddings = processor.generate_embeddings(normalized, batch_size=32, progress=True)
# 7. Slide-level aggregation
slide_embedding = processor.aggregate_embeddings(embeddings, method="mean")
print(f"Slide embedding: {slide_embedding.shape}")
# 8. Visualize
processor.plot_feature_map(normalized, embeddings, slide)Performance Considerations
When processing large WSIs, consider the following:
- CuCIM backend: Automatically preferred when available; provides GPU-accelerated slide reading
- Thumbnail-resolution detection: Run tissue detection on downsampled thumbnails to save time on initial segmentation
- Multi-resolution extraction: Build a coarse tissue grid
at low magnification, then pass it to a high-resolution extractor via
tissue_coordinates - Batch sizes: Tune
batch_sizeingenerate_embeddings()to balance GPU memory and throughput - Quality filtering before embedding: Filter out low-quality patches before the expensive embedding step to avoid wasted compute
- Immutable Patches:
filter(),normalize(), and slicing return newPatchesinstances — the original is never modified