Overview
The molecular processing pipeline in HoneyBee is backed by
SeNMo
(Self-Normalizing Multi-Omics Neural Network), a pretrained pan-cancer model
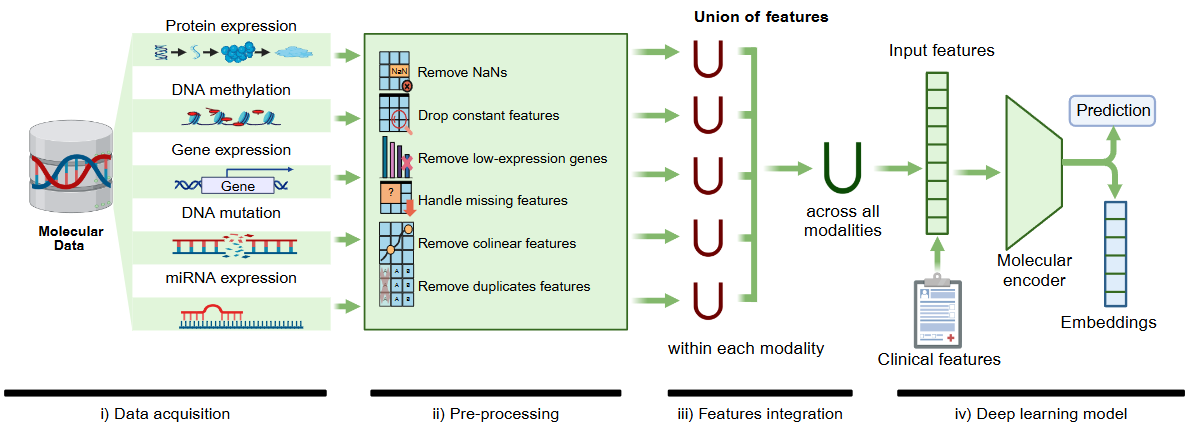
that consumes six concatenated molecular modalities — gene expression,
DNA methylation, miRNA expression, protein expression, somatic mutations, and
clinical covariates — and produces a 48-dimensional patient embedding
plus a Cox-style hazard score for survival prediction. The
MolecularProcessor class exposes the full pipeline through
HoneyBee.process_molecular() and accepts three input modes so
it slots into existing TCGA workflows without restructuring.
Key Features
-
Pretrained 10-checkpoint ensemble from
Lab-Rasool/SeNMoon HuggingFace Hub, auto-downloaded on first use -
Six per-modality preprocessing helpers ported faithfully from SeNMo's
package_classes/ - Concatenates to the published 80,697-dim feature vector; missing modalities are zero-padded for their slice
- 48-dim patient embedding and scalar hazard score per patient
- Three input modes covering raw TCGA TSVs, pre-combined pkls, and direct vectors
-
Reproducible — the DNA mutation preprocessor takes an explicit
seed(upstream'srandom.sampleis unseeded) -
High-level
HoneyBee.process_molecular()one-call API
Installation
The molecular pillar's runtime dependencies (PyTorch, NumPy, pandas) are already part of HoneyBee's core install. The 10-checkpoint ensemble (several GB) is downloaded lazily on first use and cached locally via the standard HuggingFace Hub cache.
pip install honeybee-mlQuick Start
Pass a preprocessed 80,697-dim multi-omics feature vector to
process_molecular() and get back a
MolecularResult with the embedding and hazard score:
from honeybee import HoneyBee
import numpy as np
hb = HoneyBee()
# Placeholder: in practice, build this from real per-modality TSVs (see below)
features = np.random.randn(80697).astype(np.float32)
result = hb.process_molecular(features=features)
print(f"Embedding shape: {result.embedding.shape}")
print(f"Hazard score: {result.hazard_score:.4f}")Embedding shape: (48,)
Hazard score: -0.1223
The first call downloads the 10 published SeNMo checkpoints from
HuggingFace Hub. Subsequent calls reuse the local cache. The default cache
lives at ~/.cache/huggingface/hub/ and respects the standard
HF_HOME override.
Three Input Modes
process_molecular() accepts input in any of three forms.
Exactly one of features=, features_pkl=, or
raw= must be provided.
Mode A — pre-combined pkl
Load a pickle in the format produced by SeNMo's
combine_features.py (a dict with
data['cv_splits'][1]['test']['x_omic'][0] holding the
80,697-dim vector). Useful if you've already run the upstream SeNMo
preprocessing pipeline and just want the embedding.
result = hb.process_molecular(features_pkl="multiomic_features.pkl")
print(result.embedding.shape, result.hazard_score)Mode B — raw per-modality data
Pass a dict mapping modality name to either a TSV/MAF path or a DataFrame. HoneyBee runs each per-modality preprocessor, concatenates the outputs to the 80,697-dim vector, and runs inference. Any omitted modality is zero-padded for its slice.
result = hb.process_molecular(raw={
"gene_expression": "gene-expr-RNAhtseq_fpkm.tsv",
"dna_methylation": "methylation450.tsv",
"mirna": "mirna.tsv",
"protein": "rppa.tsv",
"dna_mutation": "wxs.maf",
"clinical": "phenotype.tsv",
# Any modality may be omitted; its slice is zero-padded.
})
The seed keyword on process_molecular() (default
42) controls the DNA mutation preprocessor's random row-drop
so results are reproducible across runs. The DNA mutation source is a
standard TCGA MAF; the Hugo symbol vocabulary is fetched from the
Lab-Rasool/honeybee-samples HuggingFace dataset on first use (cached afterwards).
Mode C — preprocessed vector
Pass the 80,697-dim vector directly. Useful for batch inference where feature vectors are built offline, or for unit testing.
# Single sample
result = hb.process_molecular(features=features_80697)
# Batched (N samples)
result = hb.process_molecular(features=features_N_x_80697)
# result.embedding.shape -> (N, 48)
# result.hazard_score.shape -> (N,)Per-Modality Preprocessing
All six preprocessing helpers live under
honeybee.processors.molecular.preprocessing and accept either
a path to a TSV (or MAF, for DNA mutation) or an already-loaded DataFrame.
Each returns a pandas DataFrame in the shape
combine_modalities() expects. Function names and target
feature counts match the SeNMo paper:
| Modality | Function | Output features |
|---|---|---|
| Gene expression (RNA-seq) | preprocess_gene_expression | 8,794 |
| DNA methylation | preprocess_dna_methylation | 52,396 |
| miRNA expression | preprocess_mirna | 1,730 |
| Protein expression (RPPA) | preprocess_protein | 472 |
| DNA mutation | preprocess_dna_mutation | 17,301 |
| Clinical covariates | preprocess_clinical_covariates | 4 |
from honeybee.processors.molecular.preprocessing import (
preprocess_gene_expression,
preprocess_dna_methylation,
preprocess_mirna,
preprocess_protein,
preprocess_dna_mutation,
preprocess_clinical_covariates,
)
gene_df = preprocess_gene_expression("gene-expr-RNAhtseq_fpkm.tsv")
print(gene_df.shape) # (1, 8795) for a single sample, or (N_samples, 8795) if multiple samples are present; sample col + 8794 featuresCombine Modalities
combine_modalities() concatenates per-modality DataFrames
(or CSV paths) into the 80,697-dim SeNMo input vector. Use it if you want
to inspect or save the combined features before inference, or if your
workflow already has per-modality outputs but no upstream pkl.
from honeybee.processors.molecular.preprocessing import combine_modalities
vector = combine_modalities({
"gene_expression": gene_df,
"dna_methylation": methyl_df,
"mirna": mirna_df,
"protein": protein_df,
"dna_mutation": mutation_df,
"clinical": clinical_df,
})
print(vector.shape, vector.dtype) # (80697,) float32
# Then run inference
result = hb.process_molecular(features=vector)Result Schema
MolecularResult is a dataclass with three fields:
-
embedding— 48-dim ndarray for single-patient input, shape(N, 48)for batches. The encoder output from SeNMo's penultimate layer. -
hazard_score— Pythonfloatfor single-patient input, ndarray of shape(N,)for batches. Cox-style continuous risk score; higher means worse prognosis. -
input_features— the resolved 80,697-dim vector that was fed to SeNMo, useful for caching or downstream analysis.
Call result.to_dict() to get a JSON-serializable
representation.
About SeNMo
SeNMo is a 7-layer MLP with ELU activations and AlphaDropout (~83.3 M parameters) trained on over 10,000 patient profiles across 33 TCGA tumor types. The pretrained weights HoneyBee downloads are the 10-fold cross-validation ensemble from the published pan-cancer model. See Waqas et al. 2025 (Int. J. Mol. Sci. 26:7358) for the full architecture, training procedure, and benchmark results.